Respuestas ejercicios deber AWK
Respuestas ejercicios deber AWK¶
A continuación se presentan algunos ejercicios sobre el tema de AWK aplicados a Bioinformática.
Nota: Para todos los ejercicios asumimos que están trabajando dentro del directorio _archivos, por lo que comprueben que est se cumpla siempre que corran un comando.
ls
1. En este ejercicio se obtendrá información de un archivo multi-fasta, compuestos por varias secuencias de proteínas, en el que cada secuencia es una proteína de la bacteria Streptomyces calvuligerus. Para esto, usaremos el archivo S_clavuligerus_proteome.fasta que se encuentra en este la carpeta _archivos del repositorio de GitHub de este libro.
1.1. Determinar cuántas proteínas posee el proteoma de Streptomyces calvuligerus en base al archivo S_clavuligerus_proteome.fasta
$ gawk 'BEGIN {RS=">"} END {print NR}' S_clavuligerus_proteome.fasta
1.2. Listar los headers de las secuencias de todas las proteínas y colocar esta información en una archivo llamado headers_S_clavuligerus_proteome.txt.
$ gawk 'BEGIN {RS=">"} BEGIN {FS = "\n"} {print RS, $1}' S_clavuligerus_proteome.fasta > headers_S_clavuligerus_proteome_awk.txt
$ cat headers_S_clavuligerus_proteome_awk.txt
1.3. Obtener solamente el código Uniprot (que está entre | |, por ejemplo B5H048) de los headers de todas las secuencias de proteínas y añadir esta información en un archivo llamado codigosUniptot_S_clavuligerus_proteome.txt. Para realizar esto se puede tomar como input el archivo generado en el anterior literal.
$ gawk 'BEGIN {RS=">"} BEGIN {FS = "|"} {print $2}' headers_S_clavuligerus_proteome_awk.txt > codigosUniptot_S_clavuligerus_proteome_awk.txt
1.4. Obtener solamente el nombre de los organismos (caracteres que están luego de OS=) de los headers de todas las secuencias de proteínas y añadir esta información en un archivo llamado organismos_S_clavuligerus_proteome.txt. Para realizar esto se puede tomar como input el archivo del literal 1.3.
$ gawk 'BEGIN {RS=">"} BEGIN {FS = "="} {print $2}' headers_S_clavuligerus_proteome_awk.txt | sed -r 's/\(.+OX/ /g' > Organismos_S_clavuligerus_proteome_awk.txt
1.5. Obtener solamente el nombre de las proteínas (caracteres antes de OS=) de los headers de todas las secuencias de proteínas y añadir esta información en un archivo llamado proteinas_S_clavuligerus_proteome.txt. Para realizar esto se puede tomar como input el archivo generado en el literal 1.3.
$ gawk 'BEGIN {RS=">"} BEGIN {FS = "|"} {print $2}' headers_S_clavuligerus_proteome_awk.txt
1.6. Separar los headers y las secuencias de aminoácidos de las proteínas que tienen los siguientes códigos: B5GNS8, B5H296, B5GRF2 y B5GTR3 (para esto debes usar el archivo fasta inicial: S_clavuligerus_proteome.fasta). Colocar esta información en un archivo llamado 4seq_S_clavuligerus_proteome.fasta.
$ gawk 'BEGIN{RS=">"} /B5GNS8/ {print $0}' archivos/S_clavuligerus_proteome.fasta > archivos/4seq_S_clavuligerus_proteome_awk.fasta
$ gawk 'BEGIN{RS=">"} /B5H296/ {print $0}' archivos/S_clavuligerus_proteome.fasta >> archivos/4seq_S_clavuligerus_proteome_awk.fasta
$ gawk 'BEGIN{RS=">"} /B5GRF2/ {print $0}' archivos/S_clavuligerus_proteome.fasta >> archivos/4seq_S_clavuligerus_proteome_awk.fasta
$ gawk 'BEGIN{RS=">"} /B5GTR3/ {print $0}' archivos/S_clavuligerus_proteome.fasta >> archivos/4seq_S_clavuligerus_proteome_awk.fasta
1.7. Determinar si en la secuencia de la proteína con código Uniprot B5H296 existe un aminoácido de serina (S), seguido de una prolina (P), con tres alaninas (A) en medio de los dos aminoácidos (S y P). El patrón a buscar sería SAAAP. Puedes usar la información de la ubicación de la secuencia obtenida en el anterior literal.
$ gawk 'BEGIN{RS=">"} /B5H296/ {print $0}' S_clavuligerus_proteome.fasta | gawk '/SA{3}P/{print NR, $0}'
2. En este ejercicio se obtendrá información de archivos fastq, que contienen secuencias de nucleótidos y la calidad de su proceso de secuenciación. Para esto usaremos los archivos secuencias1.fastq, secuencias2.fastq y secuencias3.fastq que se encuentra en este la carpeta _archivos del repositorio de GitHub de este libro.
2.1. La letra N representa un nucleótido que no pudo ser leído correctamente, y se reporta como ninguno, o missing data. Imprime el número de secuencias que tenga diez N seguidas por cada uno de los archivos fastq de la carpeta _archivos.
# Opción 1
$ gawk 'BEGIN{RS="@SRR098026"} /NNNNNNNNNN/ {x++} END {print FILENAME, ": ", x}' secuencias1.fastq
$ gawk 'BEGIN{RS="@SRR098026"} /NNNNNNNNNN/ {x++} END {print FILENAME, ": ", x}' secuencias2.fastq
$ gawk 'BEGIN{RS="@SRR098026"} /NNNNNNNNNN/ {x++} END {print FILENAME, ": ", x}' secuencias3.fastq
# Opción 2
$ gawk 'BEGIN{RS="@SRR098026"} /NNNNNNNNNN/ {x++} END {print FILENAME, ": ", x}' *.fastq
2.2. Ahora, determina el número de secuencias de cada archivo .fastq por separado. Se conoce que los títulos de las corridas de las secuencias en cada archivo .fastq empiezan con el string @SRR098026.
# Opción 1
$ for f in $(ls *.fastq)
do
gawk 'BEGIN{RS="@SRR098026"} END{print FILENAME, ": ", NR}' $f
done
# Opción 2
$ gawk 'BEGIN{RS="@SRR098026"} END{print NR}' *.fastq
2.3. Pero esta información está muy desordenada. No sabemos a qué corrida le pertenece esta secuencia (identificador) ni la calidad de la secuencia con diez N seguidas. Entonces, podríamos incluir las 4 líneas de cada corrida. ¿Cómo incluirías las 4 lineas de información de cada corrida que contenga la secuencia NNNNNNNNNN del archivo secuencias1.fastq?
$ gawk 'BEGIN{RS="@SRR098026"} /NNNNNNNNNN/ {print RS, $0} ' secuencias1.fastq
2.4. Pero esta información nos podría servir mucho más si está en un archivo fastq y no solo en la pantalla del jupyter notebook o de la terminal. Qué tal si exportamos la información de todas las secuencias que contienen diez N seguidas, de todos los archivos .fastq, a un archivo llamado malas_lecturas.fastq.
$ gawk 'BEGIN{RS="@SRR098026"} /NNNNNNNNNN/ {print NR, $0}' secuencias1.fastq secuencias2.fastq secuencias3.fastq | gawk NF | sed -r '/secuencias[0-9].fastq-/d'> malas_lecturas_awk.fastq
2.5. ¿Cuántas líneas de las secuencias de cada uno de los tres archivos fastq contienen el motivo ACG?
$ gawk 'BEGIN{RS="@SRR098026"} /ACG/ {x++} END{print FILENAME, ": ", x}' *.fastq
2.6. Ahora, coloca las anteriores secuencias que contienen ACG en el archivo malas_lecturas.fastq sin sobre escribir el archivo. Luego imprime las últimas 40 líneas del archivo malas_lecturas.fastq.
$ gawk 'BEGIN{RS="@SRR098026"} /ACG/ {print NR, $0}' secuencias1.fastq secuencias2.fastq secuencias3.fastq >> malas_lecturas_awk.fastq
tail -n 40 malas_lecturas_awk.fastq
2.7. Ahora vamos a buscar patrones en el archivo malas_lecturas.fastq. Buscaremos estos motivos ACTG, CCCCC, NNNCNNN, NNNGNNN, TTTT, TATA, AAA. Coloca el resultado en el archivo acg_awk.fastq.
$ gawk '/ACTG/ || /CCCCC/ || /NNNCNNN/ || /NNNGNNN/ || /TTTT/ || /TATA/ || /AAA/ {print $0}' malas_lecturas.fastq > acg_awk.txt
2.8. Tomando el archivo acg_awk.txt, convierte los fragmentos ACG en secuencias de ARN. Recuerda que se puede hacer eso cambiando las timinas por uracilos (Reemplazar T por U).5.8. Tomando el archivo acg.txt, convierte los fragmentos ACG en secuencias de ARN. Recuerda que se puede hacer eso cambiando las timinas por uracilos (Reemplazar T por U).
$ gawk '{gsub("T", "U")} {print $0}' acg_awk.txt
3. En este ejercicio se obtendrá información de un archivo obtenido de un experimento de microarray, en el que se calcula la expresión génica de una muestra. Para esto usaremos el archivo microarray.txt que se encuentra en este la carpeta _archivos del repositorio de GitHub de este libro.
3.1. Determinar el número genes relacionados con la leucemia (en el documento se encuentra como leukemia). En este caso esto se determinará por la presencia del nombre de la enfermedad en los nombres de los genes.
$ gawk 'BEGIN {FS = "\t"} /leukemia/ {print NR}' microarray.txt | wc
3.2. De todo el registro, solo queremos el nombre y la descripción de los genes relacionados con leucemia (campos/columnas 3 y 4 del archivo), lo que se desea copiar a un archivo llamado genes_leucemia.txt.
$ gawk 'BEGIN {FS = "\t"} /leukemia/ {print $3, $4}' microarray.txt > genes_leucemia_awk.txt
$ wc genes_leucemia_awk.txt
3.3. Se desea eliminar las comillas de los nombres y descripción de los genes relacionados con leucemia, obtenidos en el archivo del literal anterior, y guardar los cambios en el mismo archivo genes_leucemia.txt.
$ gawk '{gsub("\"", "")} {print $0}' genes_leucemia_awk.txt
3.4. Ahora, se desea ordenar alfabéticamente los genes del archivo genes_leucemia.txt y eliminar los genes repetidos. Con estos cambios, guardar el resultado en un archivo llamado genes_leucemia_ord_uniq.txt
sort -u genes_leucemia.txt > genes_leucemia_ord_uniq.txt
3.5. Finalmente, se desea determinar el número de genes que estaban repetidos
# Los repetidos serían la diferencia de ambos números
wc -l genes_leucemia.txt
sort -u genes_leucemia.txt | wc -l
4. En este ejercicio se buscarán sitios d reconocimiento de enzimas de restricción en secuencias ITS de hongos. Para esto usaremos los archivos .fasta de la carpeta ITS_region que se encuentra en este la carpeta _archivos del repositorio de GitHub de este libro.
Para este ejercicio se debe conocer lo que es un mapa de restricción, que es un gráfico en el que se representan los sitios en los que diferentes enzimas de restricción poseen dianas en una molécula particular de ADN
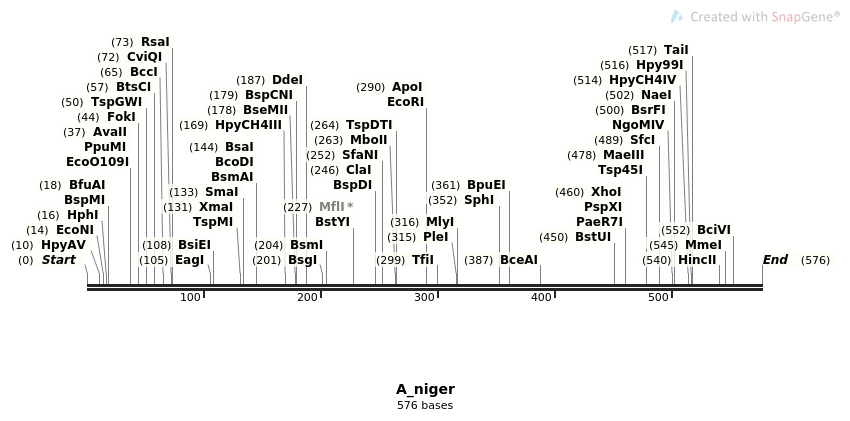
En este ejemplo se seleccionaron las siguientes enzimas de restricción:
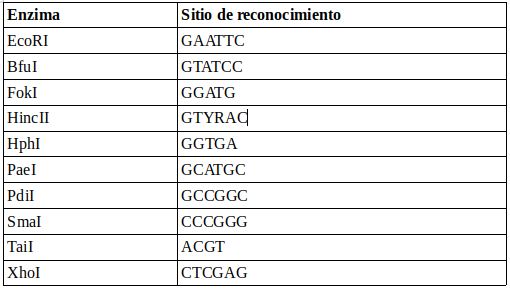
En los siguientes ejercicios se buscarán los sitios de reconocimiento de estas enzimas en la región ITS de diferentes especies de hongos.
4.1. Determinar qué archivo(s) poseen la secuencia de reconocimiento de la enzima EcoRI y cuántas de estas secuencias existen en cada uno de los archivos.
$ for f in $(ls ITS_region/*.fasta)
do
gawk 'BEGIN{RS=">"} /GAATTC/{x++} END{print FILENAME, ": ", x}' $f
done
4.2. Determinar qué archivo posee doble o triple secuencia de reconocimiento de las enzimas SmaI y TaiI.
$ for c in $(ls ITS_region/*.fasta)
do
gawk 'BEGIN{RS=">"} /CCCGGG/{x++} END{print FILENAME, ":", x}' $c
done
$ for d in $(ls ITS_region/*.fasta)
do
gawk 'BEGIN{RS=">"} /ACGT/{x++} END{print FILENAME, ":", x}' $d
done
4.3. Especificar el número de la línea donde hubo la o las coincidencias del sitio de reconocimiento de la enzima TaiI del archivo T_delbrueckii.fasta
$ gawk '/ACGT/ {print NR, $0}' ITS_region/T_delbrueckii.fasta
4.4. La enzima HincII presenta una secuencia de reconocimiento «degenerada», en la que la letra Y puede ser los nucleótidos C o T y la letra R puede ser G o A. Buscar la cantidad de estas secuencias en todos los archivos .fasta de las secuencias ITS.
$ for d in $(ls ITS_region/*.fasta)
do
gawk 'BEGIN{RS=">"} /GT[CT][AG]A/ {x++} END{print FILENAME, ":", x}' $d
done
4.5. Buscar las 10 lineas posteriores y 10 anteriores donde hubo la coincidencia de la enzima EcoRI de todas las secuencias .fasta y almcacenar los resultados en un archivo llamado sitios_rec_EcoRI.fasta. Intenta evitar imprimir el nombre del archivo del que provienen las secuencias.
$ for d in $(ls ITS_region/*.fasta)
do
gawk 'BEGIN{RS=">"} /GAATTC/{x = FNR} { if ()print }' $d
done
4.6. Verifica si la secuencia del archivo secuencia.txt está presente en el archivo secuencias.txt.
grep -f ITS_region/secuencia.txt ITS_region/secuencias.fasta
4.7. Utiliza la información del archivo sites.txt para buscar si estos sitios de restricción estań presentes en los archivos fasta de la carpeta ITS_region y cuántos existen.
grep -c -f ITS_region/sites.txt ITS_region/*.fasta
4.8. Utilizar la información del archivo sites.txt para buscar todos los sitios de restricción únicos presentes en los archivos fasta de la carpeta ITS_region
grep -h -o -f ITS_region/sites.txt ITS_region/*.fasta | sort -u