Instalación de herramientas
Contenido
Instalación de herramientas¶
Nota
Antes de comenzar con este tutorial, asegúrate de tener un compilador como gcc y librerías del lenguaje de programación C.
Para Linux debes usar los comandos:
$ sudo apt update
$ sudo apt install build-essential
$ sudo apt-get install autoconf automake make gcc perl zlib1g-dev libbz2-dev liblzma-dev libcurl4-gnutls-dev libssl-dev libncurses5-dev libncursesw5-dev
$ gcc --version
Si estás usando una MacOS instala las herramientas de línea de comandos sin instalar XCode:
$ xcode-select --install
$ gcc --version
Samtools, Bcftools, Htslib¶
Estas 3 herramientas permiten a los usuarios manipular datos producidos por métodos de secuenciación de ADN de alto rendimiento. Son librerías de funciones escritas en C que pueden utilizarse desde línea de comandos. Hay muchas opciones posibles para analizar la data. En esta unidad daremos como ejemplo un protocolo estándar de alineamiento de ADN y variant calling. Para descargar los 3 archivos ve a este url htslib. Si prefieres usar la terminal:
$ wget https://github.com/samtools/samtools/releases/download/1.12/samtools-1.12.tar.bz2
$ wget https://github.com/samtools/bcftools/releases/download/1.12/bcftools-1.12.tar.bz2
$ wget https://github.com/samtools/htslib/releases/download/1.12/htslib-1.12.tar.bz2
Nota que están comprimidas con bz2. Descompre estos archivos en tu directorio ~/Downloads y mira cómo se llaman las carpetas resultantes:
$ cd ~/Downloads
$ tar -xf samtools-1.12.tar.bz2
$ tar -xf bcftools-1.12.tar.bz2
$ tar -xf htslib-1.12.tar.bz2
$ ls -l
Si deseas puedes ver las instrucciones de instalación en la página web. De todas maneras las colocamos aquí. Mostramos el ejemplo con samtools, pero debes hacer lo mismo para el resto en una nueva carpeta dentro de /usr/local/. Por ejemplo, abajo verás que hemos instalado samtools en /usr/local/samtools. Podrías instalar bcftools en /usr/local/bcftools y htslib en /usr/local/htslib. Usaremos make, que recién aprendimos.
$ cd samtools-1.12
$ ./configure --prefix=/usr/local/samtools
$ make
$ sudo make install
Ahora hay que especificar donde se encuentra nuestro software utilizando la variable de ambiente PATH. Abre tu archivo ~/.bashrc si eres Linux o ~/.zshrc si eres MacOS con vim y escribe la ubicación de la carpeta con binarios, donde están todas las funciones. Como ejemplo lo haremos para samtools, pero debes hacerlo para las otras igualmente. Abre con vim o nano el archivo ~/.bashrc en Ubuntu o ~/.zshrc en MacOS y escribe este texto al final del archivo (lo mismo para los otros programas). Guarda antes de salir:
# SAMTOOLS
export PATH="/usr/local/samtools/bin:$PATH"
En vim se ve de esta manera.
Prueba que todo lo que instalaste funcione llamando a cada herramienta. Samtools por ejemplo mostrará este resultado. Puedes usar cualquier programa dentro del directorio /usr/local/samtools/bin. Nosotros usamos samtools.
$ ls /usr/local/samtools/bin
$ which samtools
$ samtools
Program: samtools (Tools for alignments in the SAM format)
Version: 1.12 (using htslib 1.12)
Usage: samtools <command> [options]
Commands:
-- Indexing
dict create a sequence dictionary file
faidx index/extract FASTA
fqidx index/extract FASTQ
index index alignment
...
BWA¶
BWA es un alineador de secuencias que permite tomar un genoma de referencia y mapear varias secuencias fastq. Si deseas ver las instrucciones por tu cuenta, el link es este, pero igual las colocamos resumidas aquí. Las aplicaciones son muchas, pero trataremos de usar lecturas fastq que no sean muy pesadas, como el genoma del SARS-CoV-2. La instalación de BWA es similar, lo que debes hacer es descargar el instalador, descromprimirlo, correr make y actualizar PATH. Si eres MacOS actualiza ~/.zshrc. S
$ cd /usr/local
$ sudo wget -O bwa-0.7.17.tar.bz2 https://sourceforge.net/projects/bio-bwa/files/latest/download
$ sudo tar -xf bwa-0.7.17.tar.bz2
$ sudo rm bwa-0.7.17.tar.bz2
$ cd bwa-0.7.17
$ sudo make
Abre con vim o nano el archivo ~/.bashrc en Ubuntu o ~/.zshrc en MacOS y escribe este texto (lo mismo para los otros programas). Guarda antes de salir:
# BWA PATH
export PATH="/usr/local/bwa-0.7.17:$PATH"
Ahora ejecuta esto:
$ source ~/.bashrc
$ which bwa
$ bwa
Program: bwa (alignment via Burrows-Wheeler transformation)
Version: 0.7.17-r1188
Contact: Heng Li <lh3@sanger.ac.uk>
Usage: bwa <command> [options]
Command: index index sequences in the FASTA format
...
SRA Command Line Tools¶
Sequence Read Archive (SRA) es una base de datos de secuenciación de nueva generación. La usaremos para hacer queries a la base de datos y descargar secuencias fastq. La instalación puede ser un poco larga. El sotfware es pre-compilado, entonces solo es necesario descargarlo y estará listo para usar. La documentación se encuentra aquí, pero puedes seguir los pasos resumidos de este libro también.
Si usas Linux:
$ cd /usr/local
$ sudo wget https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/2.11.0/sratoolkit.2.11.0-ubuntu64.tar.gz
$ sudo tar -xzvf sratoolkit.2.11.0-ubuntu64.tar.gz
$ sudo rm sratoolkit.2.11.0-ubuntu64.tar.gz
Ahora abre con vim o nano el archivo ~/.bashrc y escribe este texto al final del archivo. Guarda antes de salir:
# SRA TOOLKIT
export PATH="/usr/local/sratoolkit.2.11.0-ubuntu64/bin:$PATH"
Si usas MacOS:
$ cd /usr/local
$ sudo wget https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/2.11.0/sratoolkit.2.11.0-mac64.tar.gz
$ sudo tar -xzvf sratoolkit.2.11.0-mac64.tar.gz
$ sudo rm sratoolkit.2.11.0-mac64.tar.gz
Ahora abre con vim o nano el archivo ~/.zshrc y escribe este texto al final del archivo. Guarda antes de salir:
# SRA TOOLKIT
export PATH="/usr/local/sratoolkit.2.11.0-mac64/bin:$PATH"
Nota
No olvides de actualizar la Terminal
Si usas Linux:
$ source ~/.bashrc
Si usas MacOS:
$ source ~/.zshrc
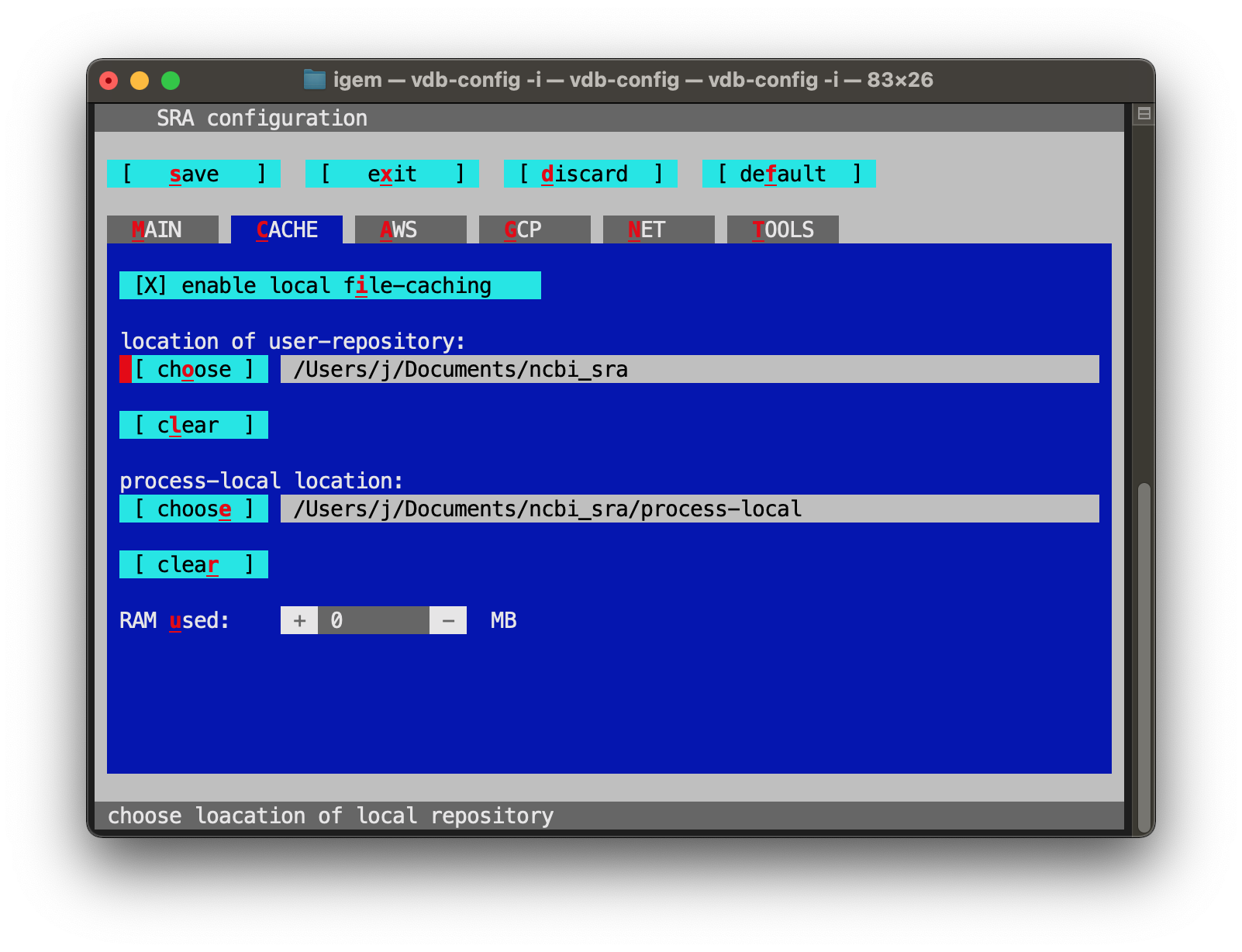
Ahora deberás configurar este software. Lo que te pedirá es ubicar el directorio de descarga para tus secuencias. Puedes ver las instrucciones de la configuración aquí, pero también recomendamos que hagas lo siguiente. Copia la dirección que sale después del comando pwd.
$ mkdir ~/Documents/ncbi_sra
$ cd ~/Documents/ncbi_sra
$ pwd
$ vdb-config -i
Ahora saldrá esta ventana. Como puedes ver debes navegar hacia la pestaña CACHE y en el campo select directory debes pegar la dirección que acabaste de copiar. Luego guarda los cambios. El campo process-local debería agregarse solo luego de agregar el primer directorio.
Ahora verifica que esté instalado.
$ fastq-dump --help
Usage: fastq-dump [ options ] [ accessions(s)... ]
Parameters:
accessions(s) list of accessions to process
Options:
...
Comentarios finales¶
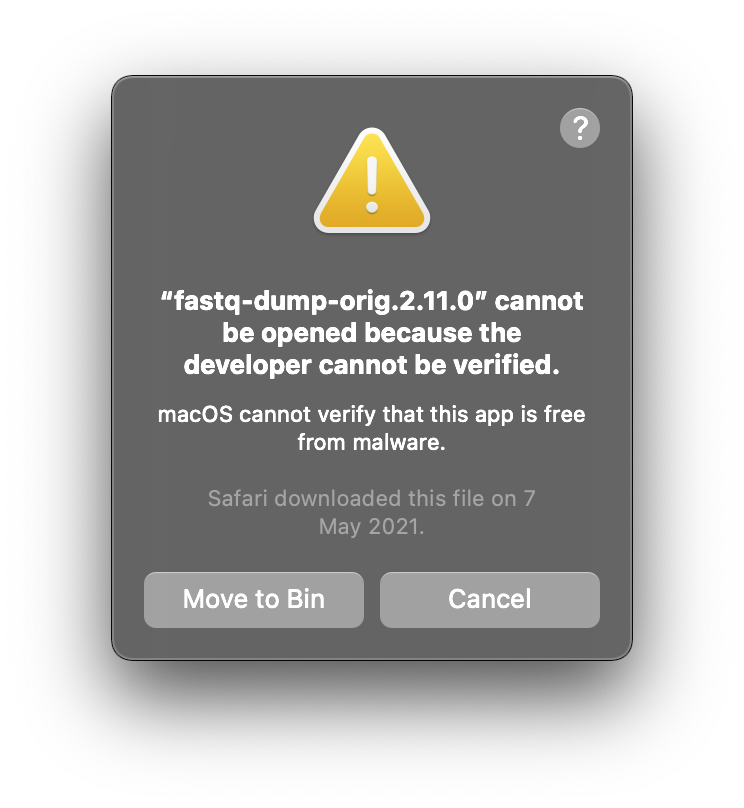
Si tienes MacOS es posible que debas usar comandos en directorios protegidos, para lo cual es necesario usar el comando sudo, como lo hicimos arriba. En Ubuntu no suele ser un problema. Luego, cuando uses los comandos en MacOS, puede ser que la computadora te pida permisos de seguridad de sistema porque MacOS es un sistema operativo bastante seguro y evita que el usuario instale malware o software de desarrolladores no autorizados oficialmente. La ventana puede lucir como esta:
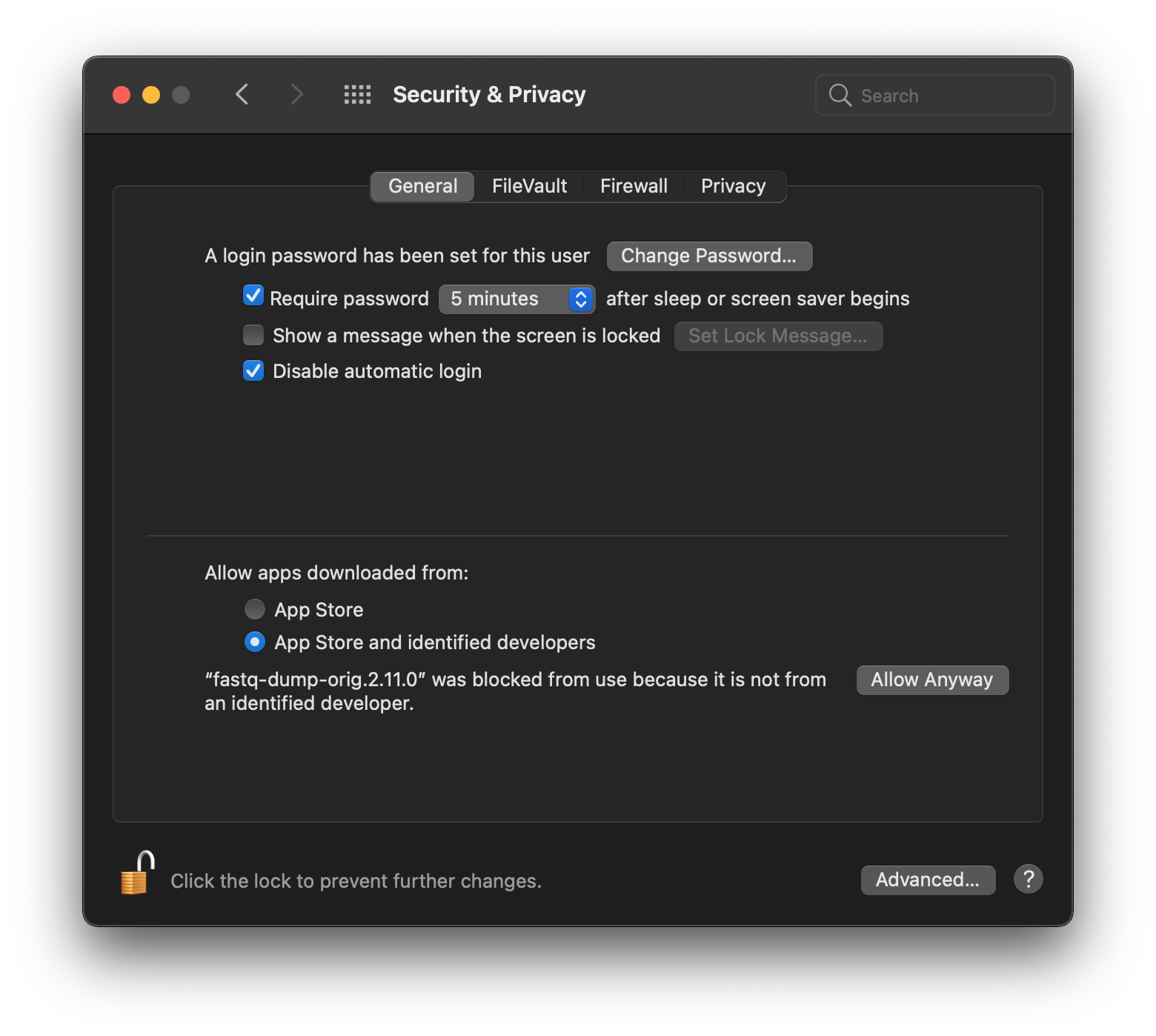
Para arreglarlo solamente debes ir a preferencias del sistema y en la pestaña de Seguridad y Privacidad, desbloquear el Firewall y permitir la ejecución del software. Probablemente debas hacerlo varias veces, pero esta bien.
¡Hemos utilizado varios de los comandos previamente estudiados como make, wget, cd, tar y echo para hacer muchas cosas sencillas! Ya entiendes mejor cómo manejar la línea de comandos.