Descarga de secuencias
Contenido
Descarga de secuencias¶
Para este ejercicio vamos a descargar el genoma de referencia del virus SARS-CoV-2 y lo usaremos como referencia para alinear secuencias de genomas de personas infectadas en Ecuador. Para ello, usaremos los comandos instalados anteriormente.
Nota
Recuerda que no debes hacer esto en un jupyter notebook, sino solo desde una ventana de Terminal.
Debes tener aún tu carpeta donde guardas tus archivos para el curso. Abrela y entra, para allí trabajar.
$ cd ~/taller_unix/7_sam_tools
Antes vamos a instalar algunos paquetes de conda para que puedas hacer queries a la base de datos NCBI.
$ conda install -n bash -c bioconda -y -c conda-forge -c bioconda -c defaults entrez-direct
$ conda activate bash
Genoma de referencia¶
El genoma de referencia reportado por NCBI se encuentra bajo el código de acceso NC_045512.2, como se puede ver en la página web oficial. La misma secuencia también se encuentra bajo el código de acceso MN908947. Este genoma tiene un ensamblaje en la base de datos Assemble de NCBI. Vamos a buscarlo con un query mediante el comando esearch. Obtendremos datos en formato XML que podremos pasar con un pipe a esummary y xtract para conocer información sobre el ensamblaje como AssemblyAccession, AssemblyStatus, SpeciesName:
$ mkdir ref_genome
$ cd ref_genome
$ echo ASSEMBLY DATA > covid_esearch.txt
$ esearch -db assembly -query '"Severe acute respiratory syndrome coronavirus 2" \
[Organism] AND (latest[filter] AND "complete genome"[filter] AND \
"reference genome"[filter] AND all[filter] NOT anomalous[filter])' | \
esummary | xtract -pattern DocumentSummary -element \
AssemblyAccession,AssemblyStatus,SpeciesName >> covid_esearch.txt
$ echo >> covid_esearch.txt
$ cat covid_esearch.txt
ASSEMBLY DATA
GCF_009858895.2 Complete Genome Severe acute respiratory syndrome-related coronavirus
...
Ahora, con ese número de acceso para el ensamblaje vamos a buscar el acceso a la secuencia del genoma RefSeq. Podemos usar el código GCF_009858895.2 directamente, pero también el comando elink para obtener el identificador único o URL al repositorio en la base de datos de NCBI.
$ echo REFSEQ GENOME DATA >> covid_esearch.txt
$ esearch -db assembly -query GCF_009858895.2 | elink -target nucleotide -name \
assembly_nuccore_refseq | esummary | xtract -pattern DocumentSummary \
-element AccessionVersion,Title >> covid_esearch.txt
$ echo >> covid_esearch.txt
$ cat covid_esearch.txt
ASSEMBLY DATA
GCF_009858895.2 Complete Genome Severe acute respiratory syndrome-related coronavirus
REFSEQ GENOME DATA
NC_045512.2 Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome
Ahora vamos a descargar la secuencia en formato .fasta con una pequeña variación al código de arriba solo utilizando efetch.
$ esearch -db assembly -query GCF_009858895.2 | elink -target nucleotide -name \
assembly_nuccore_refseq | efetch -format fasta > NC_045512.2.fasta
$ head -n 3 NC_045512.2.fasta
>NC_045512.2 Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome
ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGTAGATCTGTTCTCTAAA
CGAACTTTAAAATCTGTGTGGCTGTCACTCGGCTGCATGCTTAGTGCACTCACGCAGTATAATTAATAAC
Otra posible opción pudo ser solamente usar el código de acceso en la base de datos Nucleotide:
$ esearch -db nucleotide -query NC_045512.2 | efetch -format fasta \
> NC_045512.2.fasta
$ head -n 3 NC_045512.2.fasta
>NC_045512.2 Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome
ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGTAGATCTGTTCTCTAAA
CGAACTTTAAAATCTGTGTGGCTGTCACTCGGCTGCATGCTTAGTGCACTCACGCAGTATAATTAATAAC
Lecturas de genomas¶
Las lecturas de los genomas en formato .fastq se encuentran en la base de datos SRA de NCBI. Es importante que hayas instalado las herramientas de línea de comando para SRA, ya que así será muy fácil descargar la información. Comprueba que tengas acceso al comando prefetch desde la Terminal. Este comando permite descargar datos del repositorio SRA en formato .sra. Para transformarlos a .fastq y muchas más opciones se puede utilizar el comando fastq-dump.
$ prefetch --help
Usage: prefetch [ options ] [ accessions(s)... ]
Parameters:
accessions(s) list of accessions to process
...
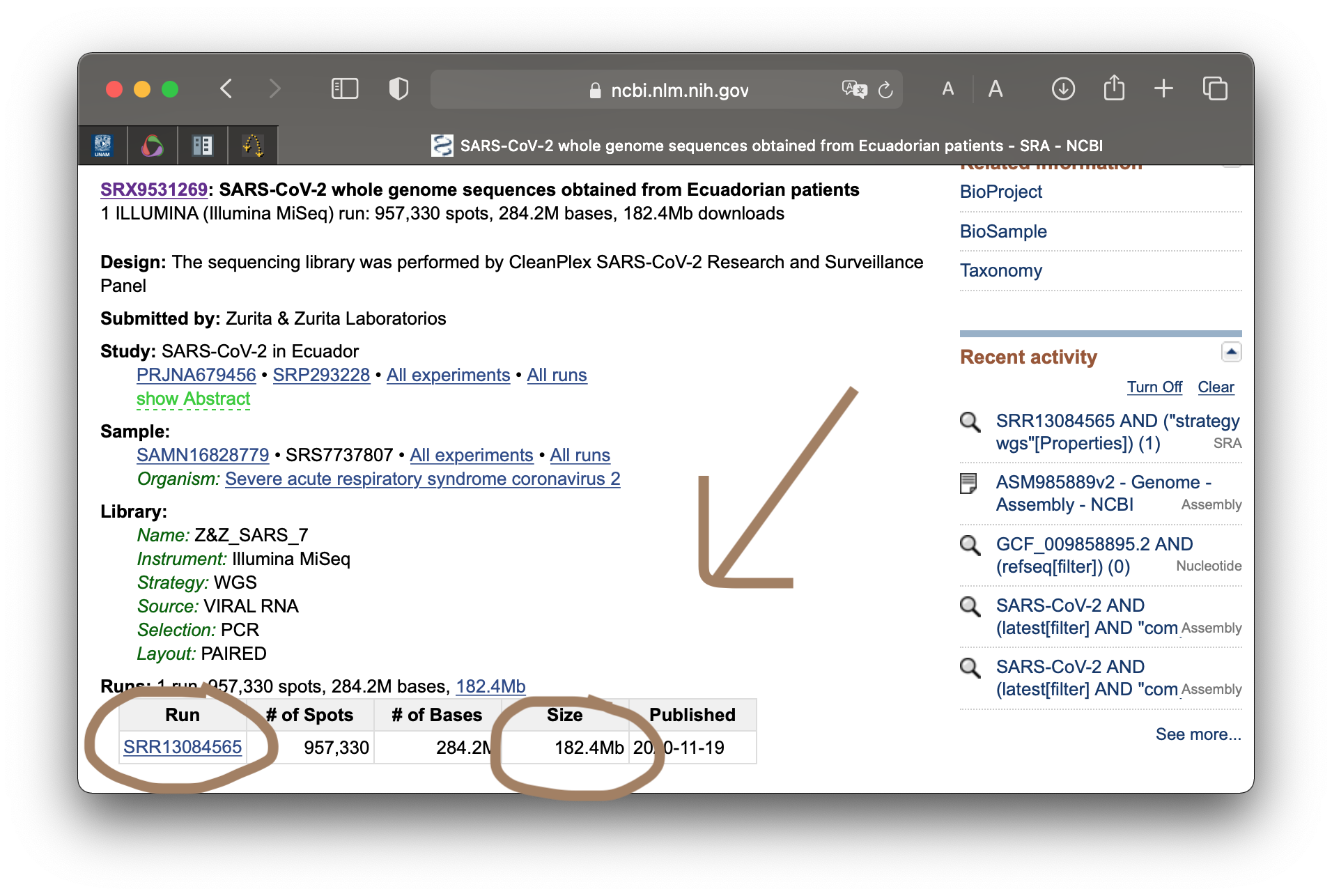
Hay muchísimas nuevas entradas a la base de datos que se podrían utilizar. Para esta ocasión decidimos utilizar secuencias que se han reportado en Ecuador, bajo el código de acceso SRR13084565. Podemos hacer un query para verificar su origen.
$ cd ../
$ esearch -db sra -query SRR13084565 | esummary | xtract -pattern DocumentSummary \
-element Title, Platform instrument_model
SARS-CoV-2 whole genome sequences obtained from Ecuadorian patients ILLUMINA
Ahora, para descargar las secuencias hay que utilizar los siguientes comandos:
$ prefetch SRR13084565
2021-05-12T12:06:07 prefetch.2.11.0: 1) Downloading 'SRR13084565'...
2021-05-12T12:06:32 prefetch.2.11.0: HTTPS download succeed
2021-05-12T12:06:33 prefetch.2.11.0: 'SRR13084565' is valid
2021-05-12T12:06:33 prefetch.2.11.0: 1) 'SRR13084565' was downloaded successfully
2021-05-12T12:06:33 prefetch.2.11.0: 'SRR13084565' has 0 unresolved dependencies
$ fastq-dump SRR13084565
Read 957330 spots for SRR13084565
Written 957330 spots for SRR13084565
$ head -n 1 SRR13084565.fastq
@SRR13084565.1 1 length=301
Es también posible hacerlo desde la página web, pero es menos divertido. Utilizamos estas lecturas porque su tamaño no es tan grande. Solo usa 184.2 Mb en la memoria. Usualmente las lecturas de nueva generación pueden contener gigas. Ahora, si deseas jugar con estas herramientas puedes usar el genoma humano y mapear lecturas del exoma, o usar el genoma de una planta. El cielo es el límite. Procura tener memoria en la computadora.
Recapitulación¶
Se descargado dos archivos desde las bases de datos de NCBI: el genoma de referencia del virus SARS-CoV-2 con código de acceso NC_045512.2 en formato .fasta y lecturas de secuenciación de nueva generación de Ecuador con código de acceso SRR13084565 en formato .fastq. Ahora utilizaremos estas secuencias para hacer un alineamiento y encontrar SNPs.