Implementación
Contenido
Implementación¶
El procesamiento de las secuencias es sencillo:
Indexar el genoma de referencia [
bwa]: Permite nombrar partes importantes que se usen como referencia en el alineamiento.Alinear las lecturas secuenciadas con el genoma de referencia [
bwa]: El propósito es claro. Hay que ubicar las secuencias obtenidas en el genoma. Este alineamiento se guarda en un archivo de formato SAM (Sequence Alignment Map), que es legible para humanos.Comprimir el archivo de alineamiento SAM [
samtools]: Convierte el archivo SAM a formato BAM (Binary Alignment Map), es un archivo binario, comprimido, que los algoritmos pueden manipular de mejor manera.Ordenar las secuencias [
samtools]: Al alinear, todo está en desorden. Se ordenan las secuencias alineadas con respecto a su ubicación en el genoma de referencia.Indexar el alineamiento [
samtools]: Permite darle un nombre a las regiones que se alinean con el genoma y luego ubicarlas fácilmente en software GUI de visualización de genomas.Obtener variaciones [
samtools]: Se obtienen las mutaciones de cada lectura del.fastainicial con respecto al genoma de referencia. Se genera un archivo.bcfObtener variantes [
bcftools]: Se obtienen los variantes del genoma de referencia. Se genera un archivo.vcf.gz.
Nota
Antes de empezar, asegúrate de que la estructura de tus archivos en tu directorio de trabajo ~/taller_unix es como la siguiente. Si prefieres usar tu propio orden, asegúrate de modificar los comandos apropiadamente.
taller_unix
├─── ...
└─── 7_sam_tools
├─── SRR13084565.fastq
└─── ref_genome
├─── covid_esearch.txt
└─── NC_045512.2.fasta
Indexar genoma de referencia¶
$ ~/taller_unix/7_sam_tools
$ bwa index ref_genome/NC_045512.2.fasta
[bwa_index] Pack FASTA... 0.00 sec
[bwa_index] Construct BWT for the packed sequence...
[bwa_index] 0.01 seconds elapse.
[bwa_index] Update BWT... 0.00 sec
[bwa_index] Pack forward-only FASTA... 0.00 sec
[bwa_index] Construct SA from BWT and Occ... 0.00 sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa index ref_genome/NC_045512.2.fasta
[main] Real time: 0.016 sec; CPU: 0.021 sec
$ ls -la ref_genome
Verás que aparece en pantalla varios archivos generados después de indexar el genoma de referencia dentro del directorio ./ref_genome. Todos estos archivos son necesarios para el alineamiento.
Alinear lecturas con genoma de referencia¶
El alineamiento puede tardar un rato. Es importante conocer que para acelerar el proceso de alineamiento es posible segmentar en sub-procesos o threads con el flag -t. En este caso hemos escogido 5. Puedes escoger más o menos, dependiendo de tu procesador. Debería funcionar con 5. Si quieres más rapidez intenta con 8. Si no funcionó intenta con 4.
$ bwa mem -t 5 ./ref_genome/NC_045512.2.fasta SRR13084565.fastq > mapa.bwa.sam
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 168426 sequences (50000554 bp)...
[M::process] read 168436 sequences (50000526 bp)...
[M::mem_process_seqs] Processed 168426 reads in 53.928 CPU sec, 17.426 real sec
[M::process] read 168436 sequences (50000126 bp)...
[M::mem_process_seqs] Processed 168436 reads in 60.813 CPU sec, 22.382 real sec
[M::process] read 168442 sequences (50000264 bp)...
[M::mem_process_seqs] Processed 168436 reads in 58.409 CPU sec, 22.926 real sec
[M::process] read 168456 sequences (50000530 bp)...
[M::mem_process_seqs] Processed 168442 reads in 67.305 CPU sec, 20.691 real sec
[M::process] read 115134 sequences (34188107 bp)...
[M::mem_process_seqs] Processed 168456 reads in 67.634 CPU sec, 22.829 real sec
[M::mem_process_seqs] Processed 115134 reads in 43.653 CPU sec, 17.589 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 5 ./ref_genome/NC_045512.2.fasta SRR13084565.fastq
[main] Real time: 124.745 sec; CPU: 352.569 sec
Podemos visualizar el archivo si usamos tail. Es un archivo con muchos datos. Quizá se vea muy desordenado. Se verán los 11 campos obligatorios de un archivo SAM.
$ tail mapa.bwa.sam
$ clear
Conversión SAM a BAM¶
Habían varias secuencias. La conversión puede tardar un rato. Podemos utilizar el comando samtools view para visualizar mapa.bwa.bam (recuerda que es un binario y si lo abres en vim o nano no vas a entender nada). Hacemos un pipe a more, para visualizarlo. Salimos con la tecla q. Luego puedes usar la función samtools flasgstat para ver cuantos alineamientos hay.
$ samtools view -bT ./ref_genome/NC_045512.2.fasta mapa.bwa.sam > mapa.bwa.bam
$ samtools view mapa.bwa.bam | more
$ samtools flagstat mapa.bwa.bam
1683291 + 0 in total (QC-passed reads + QC-failed reads)
0 + 0 secondary
725961 + 0 supplementary
0 + 0 duplicates
1681678 + 0 mapped (99.90% : N/A)
0 + 0 paired in sequencing
0 + 0 read1
0 + 0 read2
0 + 0 properly paired (N/A : N/A)
0 + 0 with itself and mate mapped
0 + 0 singletons (N/A : N/A)
0 + 0 with mate mapped to a different chr
0 + 0 with mate mapped to a different chr (mapQ>=5)
Ordenar e indexar alineamientos¶
Este paso es sencillo. Hay que crear un archivo mapa.bwa.sort.bam con todo alineado. Luego, al archivo recientemente creado lo indexamos. Se creará un archivo llamado mapa.bwa.sort.bam.bai.
$ samtools sort mapa.bwa.bam -o mapa.bwa.sort.bam
$ samtools index mapa.bwa.sort.bam
$ ls *.bai
mapa.bwa.sort.bam.bai
Identificación de Variantes¶
Ahora, lo interesante es que podemos determinar donde se encuentran las variantes para cada secuencia. Para ello usamos el comando samtools mpileup con el flag -g para producir un archivo de formato comprimido .bcf
$ samtools mpileup -g -f ./ref_genome/NC_045512.2.fasta mapa.bwa.sort.bam > map.mpileup.bcf
[mpileup] 1 samples in 1 input files
Si deseas producir un archivo que puedas visualizar en un editor de texto, puedes correr este comando para producir un archivo .vcf:
$ samtools mpileup -v -u -f ./ref_genome/NC_045512.2.fasta mapa.bwa.sort.bam > map.mpileup.vcf
[mpileup] 1 samples in 1 input files
Al abrir este archivo se podrá visualizar los datos de una manera similar a la siguiente:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT mapa.bwa.sort.bam
NC... 5 . A <*> 0 . DP=1;I16... PL 0,3,2...
NC... 6 . A <*> 0 . DP=1;I16... PL 0,3,2...
NC... 7 . G <*> 0 . DP=6;I16... PL 0,15,...
NC... 8 . G C,<*> 0 . DP=3992;... PL 0,255...
NC... 9 . T C,<*> 0 . DP=4035;... PL 0,255...
NC... 10 . T C,G,<*> 0 . DP=4035;... PL 0,255...
NC... 11 . T C,A,G 0 . DP=4038;... PL 0,255...
NC... 12 . A G,T,<*> 0 . DP=4046;... PL 0,255...
NC... 13 . T G,C,<*> 0 . DP=4079;... PL 0,255...
NC... 14 . A C,G,T 0 . DP=4084;... PL 0,255...
NC... 15 . C T,G,A 0 . DP=4138;... PL 0,255...
NC... 16 . C T,A,<*> 0 . DP=4141;... PL 0,255...
NC... 17 . T C,G,A 0 . DP=4145;... PL 0,255...
Variant Calling¶
Vamos a extraer los variantes desde el archivo .bcf producido por mpileup. Usamos el comando bcftools call con los flags -v para llamar a los variantes, -m para considerar varios alelos, --ploidy 1 para especificar ploidía 1, -O para especificar el formato de salida, z para que se comprima en .gz, -o para especificar el nombre del archivo de salida.
$ bcftools call -v -m --ploidy 1 -O z -o map.call.vcf.gz map.mpileup.bcf
$ gzcat map.call.vcf.gz
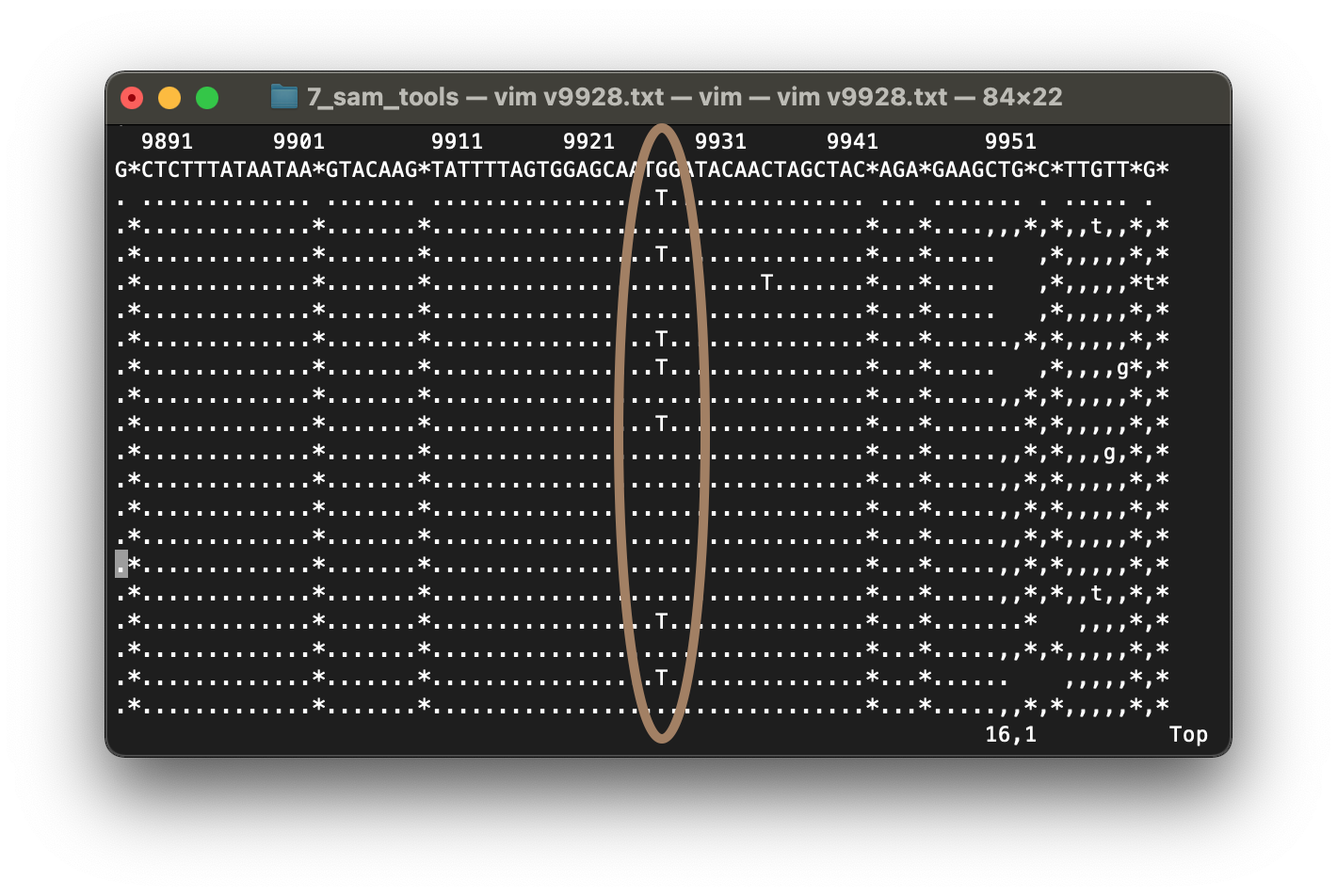
Al visualizar el resultado con gzcat vamos a obtener un formato igual al que obtuvimos antes con el archivo .vcf, pero con menos secuencias, solo con los variantes. Vamos a analizar el variante con posición 9928 (cambio de G a T) con el comando tview. Iniciamos un poco antes, en la posición 9890 y lo exportamos como archivo de texto.
$ samtools tview -p NC_045512.2:9890 -d T mapa.bwa.sort.bam ./ref_genome/NC_045512.2.fasta > v9928.txt
$ vim v9928.txt
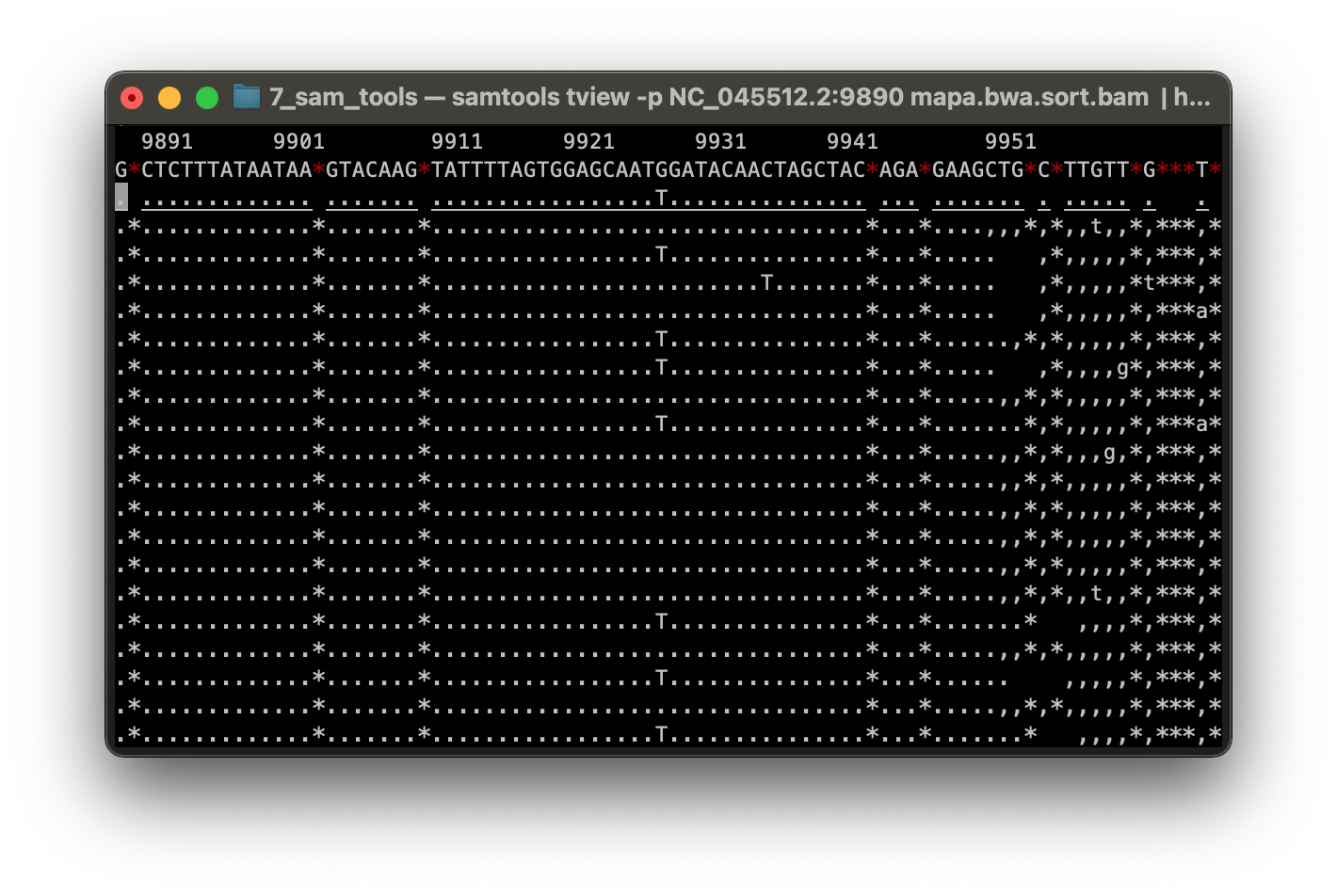
Luego visualizamos todo el alineamiento en la pantalla de Terminal.
$ samtools tview -p NC_045512.2:9890 mapa.bwa.sort.bam ./ref_genome/NC_045512.2.fasta | head
Fin¶
Las herramientas que te has instalado poseen muchas más funcionalidades. Aprender a dominar estas herramientas para procesar datos de manera eficiente es importante. En este breve taller te hemos dado algunas herramientas para el procesamiento de datos biológicos.